


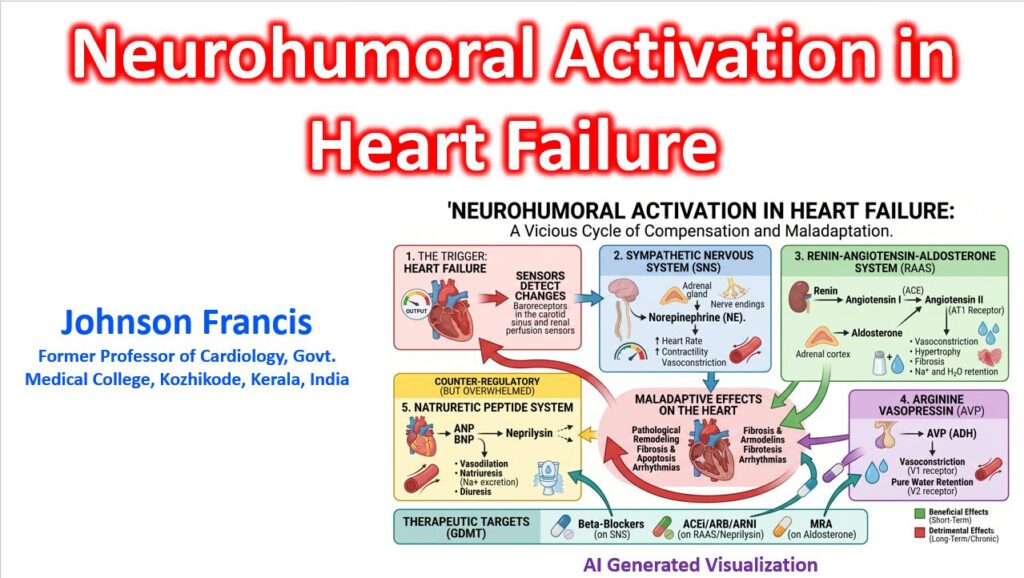

Neurohumoral activation in heart failure (HF) represents a classic physiological paradox: short-term evolutionary compensatory mechanisms—designed to maintain blood pressure and vital organ perfusion during acute volume loss—transform into the primary drivers of progressive, maladaptive disease.
When cardiac output falls, arterial underfilling unloads high-pressure baroreceptors (in the carotid sinus and aortic arch) and renal mechanoreceptors. This triggers an immediate, relentless cascade across three primary neuroendocrine axes.
The interplay between the central nervous system, failing myocardium, and circulating hormones creates a self-reinforcing loop of systemic vasoconstriction, volume retention, and direct tissue toxicity.
As the cardioregulatory center perceives diminished effective arterial blood volume, it drives sympathetic outflow and renin release, ultimately increasing cardiac workload and accelerating ventricular remodeling.
The Three Maladaptive Axes
1. The Sympathetic Nervous System (SNS)
Loss of inhibitory baroreceptor tone leads to a massive outflow of norepinephrine (NE) and epinephrine from sympathetic nerve terminals and the adrenal medulla.
- Hemodynamic impact: Increases heart rate (chronotropy) and contractility (inotropy) while causing potent peripheral vasoconstriction, driving up left ventricular afterload.
- Cellular toxicity: Chronic exposure to high circulating catecholamine levels induces myocyte apoptosis, triggers life-threatening ventricular arrhythmias, and causes a structural downregulation and uncoupling of myocardial β1-adrenergic receptors.
2. The Renin-Angiotensin-Aldosterone System (RAAS)
Renal hypoperfusion, combined with direct β1-stimulation of the juxtaglomerular apparatus, prompts the continuous release of renin.
- Angiotensin II: Acts as a potent systemic vasoconstrictor and directly stimulates myocardial and vascular hypertrophy. It also triggers the adrenal glands to release aldosterone and the posterior pituitary to secrete vasopressin.
- Aldosterone: Beyond driving distal tubular sodium and water retention, aldosterone is a potent pro-fibrotic hormone. It stimulates fibroblast proliferation and collagen deposition within the myocardial interstitium, stiffening the ventricle and worsening diastolic compliance.
3. Arginine Vasopressin (AVP)
Also known as antidiuretic hormone (ADH), AVP is released non-osmotically in response to perceived arterial underfilling.
- By binding to V1α receptors, it contributes to systemic vasoconstriction and increases afterload.
- By binding to V2 receptors in the renal collecting ducts, it drives pure free-water reabsorption. This dilutes serum sodium, making dilutional hyponatremia a classic clinical hallmark of advanced, end-stage neurohumoral activation.
The Overwhelmed Counter-Regulatory System
To counteract this profound vasoconstrictive and volume-retaining onslaught, the failing myocardium secretes Natriuretic Peptides (ANP and BNP) in response to increased wall stress and ventricular stretch.
The natriuretic peptide system attempts to promote vasodilation, natriuresis, and diuresis while directly inhibiting renin and aldosterone synthesis. However, in chronic HF, this favorable axis is completely overwhelmed by the sheer magnitude of RAAS/SNS activation. Its efficacy is further blunted by target-receptor desensitization and rapid enzymatic degradation by circulating neprilysin.
Translation to Guideline-Directed Medical Therapy (GDMT)
Modern pharmacological management of HFrEF is fundamentally an exercise in targeted neurohumoral blockade, shifting the therapeutic focus from simple hemodynamic support to interrupting these toxic cellular pathways:
| Neurohumoral Axis | Primary Pathophysiological Harm | Targeted GDMT Class |
| SNS | Catecholamine toxicity, apoptosis, arrhythmogenesis | Beta-Blockers (Carvedilol, Metoprolol succinate, Bisoprolol) |
| RAAS (Ang II) | Vasoconstriction, afterload mismatch, myocyte hypertrophy | ACEi / ARB |
| RAAS (Aldosterone) | Interstitial cardiac fibrosis, potassium wasting, volume retention | MRAs (Spironolactone, Eplerenone) |
| Natriuretic Peptides | Favorable counter-regulatory system blunted by enzymatic breakdown | ARNI (Sacubitril component inhibits neprilysin) |
By deploying an ARNI alongside a beta-blocker and an MRA, foundational therapy simultaneously severs the toxic RAAS/SNS feedback loops while artificially preserving and amplifying the heart’s endogenous protective mechanisms.
Molecular pathways of β1 adrenergic receptor downregulation and desensitization in chronic heart failure
In chronic heart failure with reduced ejection fraction (HFrEF), the continuous flood of synaptic and circulating catecholamines forces sarcolemmal β1-adrenergic receptors into a state of relentless occupancy.
Rather than allowing this hyperadrenergic state to drive unchecked calcium entry—which would trigger fatal myocyte hypercontraction, energy depletion, and necrosis—the myocyte initiates a self-protective, multi-tiered dampening cascade.
This process occurs across a strict chronological continuum: uncoupling (seconds to minutes), internalization (minutes to hours), and absolute downregulation (hours to days).
1. Receptor Uncoupling: Functional Desensitization
(Timeframe: Seconds to Minutes)
The earliest phase of desensitization blunts the receptor’s ability to transmit a signal without actually removing the receptor from the cell membrane.
- GRK2 Upregulation: The primary molecular driver is G-protein coupled receptor kinase 2 (GRK2, historically known as β-ARK1). In failing myocardium, chronic sympathetic tone drives a massive cytosolic upregulation of GRK2.
- Receptor Phosphorylation: When norepinephrine binds the β1 -AR, the receptor undergoes a conformational change that exposes its intracellular carboxyl-terminal tail and third intracellular loop. Cytosolic GRK2 identifies these active conformations and rapidly phosphorylates specific serine and threonine residues on these intracellular domains.
- β-Arrestin Recruitment: The newly attached phosphate groups act as a high-affinity molecular beacon, drawing cytosolic β-arrestin 1 and β-arrestin 2 to the membrane.
- Steric Hindrance: β-arrestin binds directly over the intracellular loops of the receptor. This creates a physical shield that blocks the receptor from interacting with its stimulatory G-protein (Gs). The Gs → Adenylyl Cyclase (AC) → cAMP → PKA (Protein Kinase A) axis is instantly severed, dropping intracellular calcium transients even though the receptor remains fully exposed to extracellular catecholamines.
2. Endosomal Sequestration: Internalization
(Timeframe: Minutes to Hours)
Once bound to the uncoupled receptor, β-arrestin shifts from a physical blocker to a structural adaptor protein, initiating the physical clearance of the receptor from the sarcolemma.
- Clathrin Lattice Formation: β-arrestin undergoes a conformational shift that exposes binding sites for clathrin and the clathrin adaptor protein 2 (AP-2) complex.
- Vesicular Invagination: The AP-2 complex gathers the uncoupled, β-arrestin-bound β1-ARs and clusters them into clathrin-coated pits on the myocyte surface.
- Dynamin Scission: The large GTPase (Guanosine Triphosphatase) enzyme dynamin wraps around the neck of the invaginated pit, constricts, and pinches it off, pulling the β1-AR inside the cell into an intracellular early endosome.
- The “Fate Fork”: Inside the endosome, the receptor faces a critical sorting decision:
- Resensitization: If the catecholamine stimulus drops, intra-endosomal phosphatases strip the phosphate groups, β-arrestin dissociates, and the receptor is recycled back to the sarcolemma.
- Degradation: In chronic HF, the unrelenting catecholamine presence forbids recycling, forcing the endosome down the degradative pathway.
3. Absolute Downregulation: Degradation & Gene Suppression
(Timeframe: Hours to Days)
Because the hyperadrenergic state of HF does not abate, the internalized receptors are permanently purged from the myocyte’s total protein pool.
- Lysosomal Destruction: The early endosomes containing sequestered β1-ARs are matured into late endosomes and trafficked to lysosomes. The vesicles fuse, and intra-lysosomal proteolytic enzymes completely degrade the receptor proteins.
- Transcriptional Suppression: Simultaneously, the prolonged activation of downstream transcription factors—specifically the Inducible cAMP Early Repressor (ICER)—binds to the promoter region of the ADRB1 (Adrenoceptor Beta 1) gene, actively halting the synthesis of new β1-AR mRNA.
- mRNA Destabilization: Post-transcriptionally, specific cytosolic RNA-binding proteins bind to the remaining ADRB1 mRNA transcripts, accelerating their enzymatic decay before they can reach the ribosomes.
The Shifting Sarcolemmal Ratio
In a healthy left ventricle, the ratio of β1 to β2 receptors is roughly 80:20. Because β2-ARs do not undergo the same aggressive GRK2-mediated internal destruction, chronic HF results in a selective loss of up to 50% to 60% of absolute β1 density. Consequently, the failing sarcolemma shifts to a severely blunted ratio of roughly 60:40.
4. Post-Receptor Collapse: The Gs-to-Gi Shift
The physical loss of β1-receptors is compounded by a profound pathological restructuring of the surviving downstream machinery:
- Upregulation of Gi: Failing myocytes explicitly upregulate the expression of inhibitory G-proteins (Gi).
- Protein Kinase A (PKA) Class-Switching: Persistent low-level PKA activity in the failing heart phosphorylates the intracellular domains of the surviving β2-receptors. This specific phosphorylation alters the receptor’s affinity, forcing it to uncouple from stimulatory Gs and couple directly to Gi.
- Active Suppression: When circulating catecholamines bind these altered β2-receptors, they now send an active inhibitory signal to adenylyl cyclase, throwing the molecular brakes on cAMP production.
Downstream Functional Consequence
Without adequate cAMP to activate Protein Kinase A, the myocyte fails to phosphorylate Phospholamban (PLN). Unphosphorylated PLN remains tightly bound to the SERCA2a pump, actively choking off the re-uptake of calcium into the sarcoplasmic reticulum.
The sarcoplasmic reticulum becomes severely depleted of calcium: systole fails because there is no calcium to release, and diastole fails because lingering cytosolic calcium prevents the myofibrils from relaxing.
The “Beta-Blocker Paradox” Explained
Understanding this precise molecular decay explains why Beta-Blockers (Carvedilol, Metoprolol succinate, Bisoprolol) serve as the cornerstone of HFrEF survival.
By acting as competitive antagonists, beta-blockers sit in the sarcolemmal binding pocket and shield the receptor from circulating catecholamines. This abruptly starves GRK2 of its target, halts β-arrestin recruitment, stops clathrin-mediated endocytosis, and drops intracellular ICER levels. Given weeks of pharmacological shielding, the myocyte resumes ADRB1 transcription, resensitizing and upregulating functional β1-receptors back to the cell surface.